


The International Commission on Non-Ionizing Radiation Protection*
Abstract
We developed a tissue culture incubator that can continuously irradiate cells with far-infrared radiation (FIR) of wavelengths between 4 and 20 μm with a peak of 7–12 μm, and found that FIR caused different inhibiting effects to five human cancer cell lines, namely A431 (vulva), HSC3 (tongue), Sa3 (gingiva), A549 (lung), and MCF7 (breast). Then, in order to make clear the control system for the effect of FIR, the gene expression concerned to the inhibition effect by FIR were analyzed. In consequence, basal expression level of HSP70A mRNA was higher in A431 and MCF7 cells than in the FIR-sensitive HSC3, Sa3, and A549 cells. Also, the over expression of HSP70 inhibited FIR-induced growth arrest in HSC3 cells, and an HSP70 siRNA inhibited the proliferation of A431 cells by irradiation with FIR. These results indicate that the effect of a body temperature range of FIR suppressing the proliferation of some cancer cells is controlled by the basal expression level of heat shock protein (HSP) 70A. This finding suggested that FIR should be very effective medical treatment for some cancer cells which have a low level of HSP70. Still more, if the level of HSP70 in any cancer of a patient was measured, the effect of medical treatment by FIR can be foreseen for the cancer. Keywords: Far-infrared radiation (FIR), CO2 incubator, Heat shock protein (HSP) 70, Human cancer cell, Over expression, siRNA
Introduction
Far-infrared radiation (FIR), which causes heating, includes electromagnetic waves with wavelengths between 4 and 1,000 μm. Recently, there have been many studies of the effects of FIR on health and in the preservation of food. The available evidence indicates that whole-body irradiation by FIR has many biological effects. For example, hyperthermia (body temperature of 39–41°C) induced by whole-body FIR has been reported to substantially inhibit spontaneous mammary tumor growth in mice [1–4]. At normal temperature ranges (approximately 25.5°C), tumor growth in SHN mice can be inhibited by FIR [5, 6]. Furthermore, whole-body FIR irradiation is believed to improve human health and sleep by enhancing blood circulation in the skin [7, 8]. This is likely due to the ability of organic matter to absorb FIR at wavelengths between 7 and 12 μm
The effects of FIR, and particularly whole-body FIR, remain unclear, because the experiments are easily affected by environmental changes in temperature and humidity and by the presence of bacteria. Therefore, we developed a chamber for raising animals that emits FIR upon heating and is capable of maintaining steady conditions. This system employs a sealed heater with a carbon/silica/aluminum oxide/titanium oxide ceramic coating produced using a polycarbonate printing technique [9]. Using this system, we found that FIR inhibits tumor growth in the A431 tumor genesis model mouse by inhibiting the expression of matrix metalloprotease-1, 9, 10, and 13. Recent in vitro studies by Teraoka et al. found that FIR at wavelengths between 4 and 16 μm inhibits the growth of HeLa cells in vitro at 37°C [10]. Despite these findings, the molecular mechanism by which FIR affects cellular gene expression remains unclear.
The lack of data on the effects of FIR on cells is due to the difficulty in stably irradiating cells with FIR under ideal culture conditions (i.e., 100% humidity, 37.0 ± 0.5°C, 5% CO2) and examining the effects of FIR at the cellular level. Therefore, using a polycarbonate printing technique, we developed a CO2 incubator with a sealed heater that has a carbon/silica/aluminum oxide/titanium oxide ceramic coating and emits FIR upon heating [11]. This CO2 incubator can stably emit FIR at wavelengths between 4 and 20 μm (maximum at 7– 12 μm) under conditions of 100% humidity, 37.0 ± 0.5°C, and 5% CO2. Our results using this incubator indicate that a body temperature range of FIR radiation suppresses the proliferation of HSC3, Sa3, and A549 cells. But, A431and MCF7 was not affected. These differences may be caused by any control system competing with FIR
Therefore, the gene expression concerning to the effect of inhibition by FIR are analyzed in the present study. Still more, the effect of over expressing and suppressing candidate FIR response genes HSP70 we examined.
Materials and methods
IFIR incubator
As previously reported [11], we fabricated an FIR radiant-panel incubator by coating a carbon/silica/aluminum oxide/titanium oxide ceramic (radiation efficiency > 97%) using a polycarbonate printing technique (Bloodissue Co. Ltd. Tokushima, Japan). The incubator has a stably irradiate system with FIR at wavelengths between 4 and 20 μm (maximum at 7–12 μm) under conditions of 100% humidity, 37.0 ± 0.5°C, and 5% CO2 in air.
Calculation of FIR absorbed per 10-cm tissue culture dish in the FIR incubator
Fourier Transform Infrared Spectroscopy (FTIR) analysis revealed that the ceramics coating inside the CO2 incubator emits FIR at 4 W m−1 str−1 μm−1 at wavelengths between 4.486 and 20.256 μm, with a maximal emission of 11.6 W m−1 str−1 μm−1 at 9 μm, which is >95% of the emission rate of an ideal black body. Since the ceramic coating was maintained at 40°C, the total generating energy, integrated over the entire range of wavelengths, was calculated to be 130.225 Wm−2 str−1. The total area of the FIR-emitting ceramic surface was 1.2385 (m2). Therefore, the total energy emitted into the incubator was 161.28366 W/str. Assuming that FIR is emitted in all directions, the total emission was 2026.7502 J/sec. Given that the volume of the CO2 incubator was 0.1257 m3 and the volume of culture medium was 6 ml, the amount of energy absorbed by each 10-cm culture dish was 0.09674 J/sec. The surface area of each 10-cm culture dish was 78.5 cm2, so that the energy reaching the base of the dish was 0.001232 J/sec/cm2. Thus, over a 1-h period 4.4352 J/h cm2 was absorbed by each 10-cm culture dish.
Cell lines and cell culture
AA431 human epithelial vulva carcinoma cells and Sa3 human gingival squamous carcinoma cells were purchased from RIKEN Cell Bank (Tsukuba, Japan). A HSC3 human tongue squamous carcinoma cells, A549 human lung carcinoma cells, and MCF7 human breast carcinoma cells were purchased from Health Science Research Resources Bank (Sennan, Japan). A431, A549, and MCF7 cells were cultured in Dulbecco’s modified Eagle’s medium/Ham’s F-12 nutrient mixture (Sigma, St. Louis, MO, USA). HSC3 and Sa3 cells were cultured in Eagle’s basal medium (Sigma). All culture medium was supplemented with 10% heat-inactivated fetal bovine serum, 100 μg/ml penicillin G, 100 μg/ml streptomycin sulfate, and 250 ng/ml amphotericin B (Invitrogen, Carlsbad, CA, USA). Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in air. The medium were replaced every 2 days.
Measurement of cell number and growth
Cells (5 × 104) were plated in triplicate in 24-well plates (Nunc, Roskilde, Denmark). The attached cell populations were measured on day 8 using 0.2% Trypan blue and a hemocytometer. Incorporation of 5-bromo-2′-deoxyuridine (BrdU) was used to determine the amount of DNA synthesis. DNA synthesis by proliferating cells was assessed using a BrdU labeling and detection kit III (Roche, Mannheim, Germany) according to the manufacturer’s protocol. Briefly, cells (5 × 103 per well) were seeded in 96-well tissue culture plates (Nunc) and then placed in the FIR incubator for 4 days, and BrdU incorporation was measured during the logarithmic growth phase (i.e., before the cells were confluent) by treating the cells for 4 h at 37°C with 10 μM BrdU. BrdU incorporation was quantified by measuring the absorbance of the substrate reaction (405 nm) and the absorbance at the reference wavelength (590 nm) using an ImmunoMini NJ-2300 (System Instruments, Tokyo, Japan). Absorbance values directly correlated with the amount of DNA synthesis and therefore the number of proliferating cells.
Histochemistry
Cells were grown on 22-mm2 glass coverslips in 6-well culture dishes (Nunc). After 4 days of FIR irradiation, the cells were observed with a CK40 phase contrast microscope (Olympus, Tokyo, Japan), fixed, and stained with hematoxylin and eosin. For immunofluorescent staining of heat shock protein (HSP) 70, cells were washed in Phosphate Buffered Saline (PBS), fixed for 20 min in 4% paraformaldehyde in PBS, washed three times for 5 min each in PBS, and blocked for 1 h at room temperature with 5% goat serum. Cells were incubated at 4°C overnight in 1:200 mouse monoclonal antibody to HSP70 (Stressgen, Victoria, Canada) in PBS containing 1 mg/ml bovine serum albumin. After washing, the cells were incubated with 1:400 FITC (Fluorescein isothiocyanate)-labeled goat anti mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The localization of intracellular HSP70 protein was identified using a BX51 confocal microscope (Olympus) and a Cool SNAP CF digital camera (Roper Scientific, Trenton, NJ, USA) and calibrated using RS Image Express software (Roper Scientific).
Microarray studies and data analysis
Four days after FIR irradiation, two control and two FIR-irradiated samples were prepared for microarray hybridization. Total RNA was extracted using a Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. Agilent human 1A ver.2 microarray slides (Agilent Technologies, Palo Alto, CA, USA) were used for the hybridization. The quality of RNA samples was monitored using an Agilent 2100 bioanalyzer (200 ng each). To produce labeled cRNA (complementary RNA), high-quality RNA was amplified and labeled with Cy5-and Cy3-CTP (Amersham Biosciences, Buckinghamshire, UK) using a Low RNA Input Fluorescent Linear Amplification Kit (Agilent) according to the manufacturer’s protocol. After the amplification and labeling, the dye incorporation ratio was determined using a Nanodrop spectrophotometer, and the ratios were within 10–20 pmol per μg cRNA, which is the range suggested by the manufacturer for hybridization. For hybridization, an Agilent 60-mer oligo microarray (Rev. 7, SSC Wash/6-screw hybridization chamber) was used according to the manufacturer’s protocol. Briefly, 750 ng Cy3-labeled control and 750 ng Cy5-labeled MPP+-treated sample were mixed and incubated for 17 h with an SSC- washed microarray slide from an Agilent In situ Hybridization Kit. Sample pairs were dye-swapped and processed at the same time. The washed slides were immediately dried under a stream of ultrapure N2 in an ozone-free atmosphere. After drying, the slides were scanned using an Agilent Technologies Microarray Scanner with the PMT setting at 770 for Cy5 and 670 for Cy3, and the raw data were normalized and analyzed using GeneSpring 7.0 software (Silicon Genetics, Santa Clara, CA, USA). For normalization, per spot and per chip intensity-dependent (LOWESS) normalization was used to correct for the intensity-dependent ratio bias12. In addition, the following filters were applied to improve the quality of the data: eliminate saturated signal, eliminate non uniformity of background, eliminate non uniformity of feature, Feature Population Outlier, eliminate low-signal feature of background signal + 2.6 × SD, and eliminate P-value < 0.01. Genes were further classified for process and function according to their GO term information (http://www.godatabase.org).
Stable transfection of HSP70A
The HSP70A expression vector pcDNA3-HSP70A containing the cDNA for full-length human HSP70A was a generous gift from Dr. Hector R. Wong (Children’s Hospital Medical Center, Cincinnati, OH, USA). HSP70A cDNA was subcloned into the Xba I and Bam HI sites of pcDNA3.1(−) (Invitrogen). Cells grown on 60-mm dishes were transfected with 8 μg of pcDNA3- HSP70A, or pcDNA3.1 (Invitrogen) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The transfected cells were selected with 400 μg/ml G418 (Sigma), and clones formed were collected and maintained separately in medium supplemented with 400 μg/ml G418.
Reverse transcription (RT)-polymerase chain reaction (PCR)
Total RNA was extracted using Trizol reagent (Invitrogen) following the manufacturer’s instructions. The concentration and purity of the RNA were determined spectrophotometrically. One microgram of total RNA was reverse- transcribed into first-strand cDNA. Next, oligo(dT12–18) primer (Invitrogen), 10 mM dNTP mix (Invitrogen), 25 mM MgCl2 (Promega, Madison WI, USA), and 0.1 M dithiothreitol (Invitrogen) were added, and the mixture was incubated for 2 min at 42°C. Next, the RT reaction was performed by adding SuperScript II RT (Invitrogen) and incubating at 42°C for 50 min, followed by 70°C for 15 min. The reaction was terminated by rapid cooling on ice, after which RNA in the sample was degraded by treatment with RNase H (Invitrogen) at 37°C for 20 min. Polymerase chain reaction (PCR) was carried out using a 1-μl sample of the RT reaction and Ready Mix PCR Master Mix (AB gene, Epsom, Surrey, UK). The analyzed genes and the respective primer sequences were as follows: HSP70A, 5′-TGTTCCGTTTCCAGCCCCCAA-3′ (sense) and 5′-GGGCTTGTCTCCGTCGTTGAT-3′ (antisense); HSP70B, 5′- CTCCAGCATCCGACAAGAAGC-3′ (sense) and 5′- ACGGTGTTGTGGGGGTTCAGG-3′ (antisense); HSP70C, 5′- TTGAGGAGGTGGATTAGGGGC-3′ (sense) and 5′-AGCCTTTGTAGTGTTTTCGCC-3′ (antisense); glyceraldehyde-3-phosphate dehydrogenase (G3PDH), 5′-ACCACAGTCCATGCCATCAC-3′ (sense) and 5′-TCCACCACCCTGTTGCTGTA-3′ (antisense). PCR was carried out using cycles (30 for HSPs and 23 for G3PDH) of 94°C for 45 s, 58°C for 30 s (all HSPs) or 52°C (G3PDH), and 72°C for 90 s. Finally, primer extension was performed for 10 min at 72°C. A 10-μl sample of each PCR product was separated by electrophoresis on a 1.5% polyacrylamide gel in Tris borate/EDTA buffer and stained with ethidium bromide.
Quantitative real-time RT-PCR data analysis
To determine the level of HSP70A mRNA, quantitative real-time RT-PCR was carried out using a LightCycler and the Fast Start DNA Master SYBR Green I Kit (Roche). The reaction contained 50 ng of cDNA and 100 pmol of each primer in a final volume of 10 μl. The gene-specific primers were as follows: HSP70A, 5′-TGTTCCGTTTCCAGCCCCCAA-3′ (sense) and 5′- GGGCTTGTCTCCGTCGTTGAT-3′ (antisense); and α-actin, 5′- ATAGCACAGCCTGGATAGCAACGTAC-3′ (sense) and 5′- CACCTTCTACAATGAGCTGCGTGTG-3′ (antisense). The concentration of Mg2+ was 3 mM. In all cases, a first phase of denaturation was performed at 95°C for 10 min. Amplification was carried out for cycles of denaturation at 95°C for 10 s, hybridization for 10 s (58°C for HSP70A or 60°C for α-actin), and elongation at 72°C (20 s for HSP70A or 10 s for α-actin). Product specificity was evaluated by melting curve analysis. Fluorescence data were analyzed using LightCycler Software Ver. 3.5 (Roche). Crossing points were established using the second derivative method. The relative amount of target transcript in the sample was calculated by dividing the amount of target by the amount of internal standard (α-actin). Results were expressed as the target/internal standard concentration ratio calculated from the calibration curve. Since the target and internal standard genes had different sequence and amplicon lengths, it was expected that they would show different PCR efficiencies. Therefore, the PCR efficiency (10−1/m, where m is the slope from the calibration curve) was first established for each pair of primers. All reactions were performed in triplicate.
HSP70 Enzyme-Linked Immuno Sorbent Assay (ELISA)
HSP70 protein was quantified in cell lysates using a commercially available ELISA kit for human HSP70 (Stressgen). Cells (105 per well) were seeded in 6-well plates (Nunc). After 4 days, the cells were lysed, and all samples were assayed at optimal dilutions according to the manufacturer’s instructions.
Protein extraction and Western blotting
Cells (1 × 106) were grown in 60-mm tissue culture dishes (Nunc). After removing the cell culture medium from the culture dishes (Nunc) and washing the cells twice with cold PBS(−), the cells were lysed in lysis buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/mL leupeptin). Protein levels were measured by the Lowry method [13] using a DC Protein Assay Kit (Bio-Rad, Hercules, CA, USA). Cell lysate containing 15 μg of protein for HSP70 was subjected to Sodium Dodesyl Sulfate (SDS)-polyacrylamide gel electrophoresis. Separated proteins were then transferred from the gel to a polyvinylidene difluoride membrane. After blocking with 5% skim milk in PBS-Tween, the membrane was incubated for 1 h at room temperature with primary antibody in PBS-T containing 5% skim milk, followed by three 10-min washes with PBS-T. Next, the membranes were incubated for 1 h at room temperature with horseradish peroxidase-labeled secondary antibody and washed three times for 10 min with PBS-T. Immunoreactive protein was detected using an ECL plus kit (Amersham Biosciences) and visualization by exposure to Hyperfilm (Amersham Biosciences). The primary antibodies used were rabbit anti-HSP70 (Stressgen), anti α-actin (Sigma), and the secondary antibody was horseradish peroxidase- conjugated anti-mouse (Zymed Laboratories, South San Francisco, CA, USA) or anti-rabbit IgG (Amersham Biosciences).
Small interfering RNA (siRNA)
We designed 21-nucleotide siRNAs targeting human HSP70A and HSP70C according to the manufacturer’s instructions (Dharmacon, Lafayette, CO, USA) and corresponding to the sequence 5′-AAGAACCAGGUGGCGCUGAAC-3′. It was not possible to design a siRNA specific to HSP70A because its mRNA was highly homologous to the mRNA for HSP70C (see Table 1). We used siCONTROL Non-Targeting siRNA (Dharmacon; 5′- UAGCGACUAAACACAUCAAUU-3′) as a negative control because it does not match the sequence of any known human or mouse genes. Cells (1 × 104/well) were plated in 96-well culture dishes (Nunc) and cultured for 24 h at 37°C in a 5% CO2 atmosphere. When the cells reached 70–90% confluence, they were transfected with 100 nM siRNAs to HSP70A and HSP70C or control siRNAs complexed with Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. The growth medium was removed after 6 h, and the culture dish was transferred to the FIR incubator. After incubation for 48 h at 37°C in a 5% CO2 atmosphere, cell proliferation was assessed by BrdU incorporation.
Table 1
Characterization of Hsp70 (A, B, and C)
| Gene | Location | Amino acid homology to HSP70 (%) | mRNA homology to Hsp70A (%) | Alternative names | GenBank |
|---|---|---|---|---|---|
| Hsp70A | n6p21.3 | 100 | 100 | HSPA1A, Hsp72, Hsp70-1a | M11717 |
| Hsp70B | lq23 | 84 | 84 | HSPA6, Hsp70B | X5175 |
| Hsp70C | 6p21.3 | 99 | 98 | HSPA1B, Hsp72, Hsp70-1b | M5980 |
Statistical analysis
Data are means ± SE of replicate samples in single experiments or replicate experiments as described in the figure legends. Student’s t-test was used for comparisons between two groups. Multiple group comparisons were performed by one-way ANOVA, followed by the Tukey–Kramer multiple group comparisons test. All statistical analyses were performed using Statcel 2 software (OMS publishing, Saitama, Japan).
Results
FIR irradiation selectively inhibits the growth of specific cancer cell lines
To clarify the effect of FIR irradiation on the proliferation of cancer cell lines, we irradiated five cancer cell lines (A431, HSC3, Sa3, A549, and MCF7) with FIR and measured the number of live cells by Trypan blue dye exclusion. Although the proliferation of HSC3, Sa3, and A549 cells were significantly suppressed on day 8 of culture (45.75%, 74.63%, and 65.79%, respectively), FIR irradiation had little effect on the growth of A431, or MCF7 cells (Fig. 1).
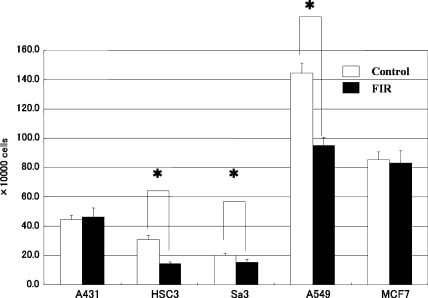 Fig. 1
Effect of FIR irradiation on cell growth of five cancer cell lines. Cells (1 × 105) were plated in 24-well dishes and cultured for 8 days. Cell numbers were counted every other day. Although proliferation of HSC3, Sa3, and A549 cells was suppressed on day 8 of culture, FIR irradiation had little effect on the proliferation of A431 and MCF7 cells. *P < 0.05 vs. unirradiated control cells
Fig. 1
Effect of FIR irradiation on cell growth of five cancer cell lines. Cells (1 × 105) were plated in 24-well dishes and cultured for 8 days. Cell numbers were counted every other day. Although proliferation of HSC3, Sa3, and A549 cells was suppressed on day 8 of culture, FIR irradiation had little effect on the proliferation of A431 and MCF7 cells. *P < 0.05 vs. unirradiated control cells
Microarray analysis and extraction of candidate gene for FIR control
Several gene families showed high correlations between endogenous expression (signal in the microarray) and the growth rate, proteins involved in cell proliferation, cytoskeletal proteins, cell cycle components, and protein kinases. Especially, in the analysis of stress factor, some genes encoding the HSPs which were well known that they participate in the cellular resistance to stress were focused. Of the 35 HSP genes on the microarray, HSP70 showed the highest correlation with the growth rate (Fig. 2a, b). We examined this further by real time RT-PCR, and we found that HSP70 is most highly expressed in A431 cells at all stages of the cell cycle. Lower levels of expression were found in HSC3 and Sa3 cells (Fig. 2c). We did not find a statistically significant difference between the expression of this gene between control (unirradiated) and FIR-irradiated A431, HSC3, or Sa3 cells. An ELISA for HSP70 in untreated cells showed similar results, specifically, that the expression of HSP70A was higher in A431 cells than in the HSC3 or Sa3 cells (Fig. 2d).
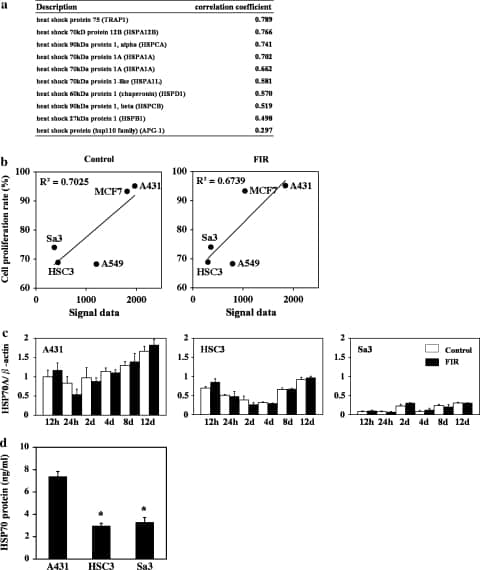 Fig. 2
Open in a separate window
Relationship between the expression of HSP70 and the suppression of proliferation by FIR irradiation. (a) Correlation between the signal from the microarray for the HSP family members in the control group and the correlation coefficient. (b) Correlation between the signal from the microarray for HSP70 and the growth rate in the control and the FIR-treated cells. (c) Expression of HSP70A mRNA in A431, HSC3, and Sa3 cells after 0.5, 1, 2, 4, 8, and 12 days of culture. The level of these mRNAs was determined by quantitative real-time RT-PCR. Bars represent the means and error bars the SD from three independent experiments. (d) Level of HSP70 protein on day 4 of culture. The concentration of HSP70 protein was measured by ELISA. Bars represent the mean, and error bars the SD (n = 4). *P < 0.01 vs. to A431 cells
Fig. 2
Open in a separate window
Relationship between the expression of HSP70 and the suppression of proliferation by FIR irradiation. (a) Correlation between the signal from the microarray for the HSP family members in the control group and the correlation coefficient. (b) Correlation between the signal from the microarray for HSP70 and the growth rate in the control and the FIR-treated cells. (c) Expression of HSP70A mRNA in A431, HSC3, and Sa3 cells after 0.5, 1, 2, 4, 8, and 12 days of culture. The level of these mRNAs was determined by quantitative real-time RT-PCR. Bars represent the means and error bars the SD from three independent experiments. (d) Level of HSP70 protein on day 4 of culture. The concentration of HSP70 protein was measured by ELISA. Bars represent the mean, and error bars the SD (n = 4). *P < 0.01 vs. to A431 cells
Increased expression of HSP70A improves the survival of HSC3 cells after a limited exposure to FIR
To directly determine whether HSP70 can protect cells from FIR-induced cell death, we developed A431 and HSC3 cell lines stably expressing human HSP70A (A431-HSP70A and HSC3-HSP70A cells, respectively). Control cells were transfected with empty pcDNA3.1 (A431-Neo and HSC3-Neo; Fig. 3a, b). In our initial experiments, we found that exposure of HSC3 and Sa3 cells but not A431 cells to limited FIR causes G2/M arrest and induces partial hypertrophy to necrosis (data not shown). To determine whether increased expression of HSP70A confers protection against FIR, cell survival was examined in FIR-irradiated A431-HSP70, A431-Neo, A431-wt, and HSC3- HSP70, HSC3-Neo, and HSC3-wt cells. We found that over expression of HSP70A increased cell proliferation in A431 and HSC3 cells. Furthermore, the proliferation of FIR-irradiated and control (unirradiated) A431-HSP70A cells was similar (Fig. 3c). The survival rate after 6 days of FIR irradiation was significantly higher in HSC3-HSP70A cells than in HSC3-Neo or HSC3-wt cells. In addition, the proliferation of FIR-treated HSC3-HSP70A cells was similar to that of control HSC3-HSP70A cells. BrdU incorporation was significantly higher in FIR-irradiated or control A431-HSP70A cells than in A431-Neo or A431-wt cells (Fig. 3d). Although BrdU incorporation of FIR- irradiated HSC3-wt and HSC3-Neo cells was lower than in unirradiated HSC3- wt and HSC3-Neo cells, it was similar in FIR-irradiated and unirradiated HSC3-HSP70A cells. Surprisingly, BrdU incorporation by HSC3-HSP70A cells was significantly higher in both FIR-irradiated and unirradiated cells. These data demonstrate that over expression of HSP70A in HSC3 and A431 cells did not affect their proliferation, and also their morphology, even when they were irradiated with FIR (Fig. 3e).
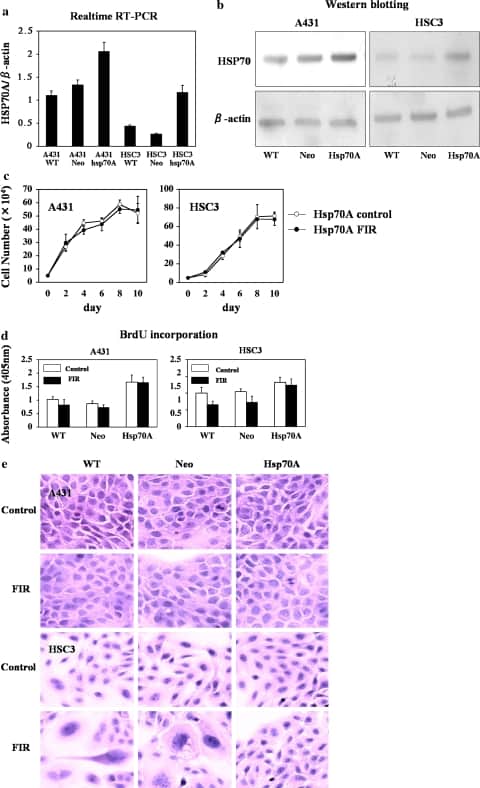 Fig. 3
Open in a separate window
Over expression of HSP70 prevents the suppression of cell growth and induction of cell hypertrophy by FIR. (a) Real-time RT-PCR of HSP70 expression. Cells overexpressing HSP70 were established. (b) Representative Western blots demonstrating increased expression of HSP70 in stably transfected A431 and HSC3 cells. Lane 1, wild-type cells; lane 2, cells transfected with empty pcDNA3.1(+); lane 3, cells transfected pcDNA3.1- HSP70A. (c) Cell proliferation of empty vector-transfected and HSP70- overexpressing cells. Cell proliferation was the same in FIR-irradiated and untreated A431 cells overexpressing HSP70. In contrast, in HSC3 cells, over expression of HSP70 prevented FIR inhibition of cell proliferation. (d) BrdU incorporation in wild-type, empty vector-transfected, and HSP70- overexpressing cells treated with FIR. (e) Hematoxylin and eosin staining of wild-type, empty vector-transfected, and HSP70A-overexpressing A431 and HSC3 cells after 5 days of transfection. Note that the morphology of HSP70A- transfected HSC3 cells was unaffected by FIR irradiation
Fig. 3
Open in a separate window
Over expression of HSP70 prevents the suppression of cell growth and induction of cell hypertrophy by FIR. (a) Real-time RT-PCR of HSP70 expression. Cells overexpressing HSP70 were established. (b) Representative Western blots demonstrating increased expression of HSP70 in stably transfected A431 and HSC3 cells. Lane 1, wild-type cells; lane 2, cells transfected with empty pcDNA3.1(+); lane 3, cells transfected pcDNA3.1- HSP70A. (c) Cell proliferation of empty vector-transfected and HSP70- overexpressing cells. Cell proliferation was the same in FIR-irradiated and untreated A431 cells overexpressing HSP70. In contrast, in HSC3 cells, over expression of HSP70 prevented FIR inhibition of cell proliferation. (d) BrdU incorporation in wild-type, empty vector-transfected, and HSP70- overexpressing cells treated with FIR. (e) Hematoxylin and eosin staining of wild-type, empty vector-transfected, and HSP70A-overexpressing A431 and HSC3 cells after 5 days of transfection. Note that the morphology of HSP70A- transfected HSC3 cells was unaffected by FIR irradiation
Knocking down HSP70A by using siRNA improves the deth of HSC3 cells after a limited exposure to FIR
We next examined the effect of knocking down HSP70A and HSP70C mRNA and HSP70 protein expression using siRNA. Transfection with HSP70A/C siRNA effectively decreased HSP70A and HSP70C mRNA (Fig. 4a) and protein levels (Fig. 4b) in both A431 and HSC3 cells without affecting the level of HSP70B mRNA or protein. HSP70A/C siRNA did not suppress BrdU incorporation in unirradiated A431 cells, but it suppressed BrdU incorporation in cells irradiated with FIR (Fig. 4c). Similarly, the HSP70A/C siRNA enhanced the suppression of BrdU incorporation by FIR irradiation. FIR irradiation also significantly suppressed BrdU incorporation in HSC3 cells transfected with the negative control siRNA (Fig. 4c). These results indicate that a decrease in HSP70 protein mediates the ability of limited FIR to inhibit the proliferation of A431 and HSC3 cells.
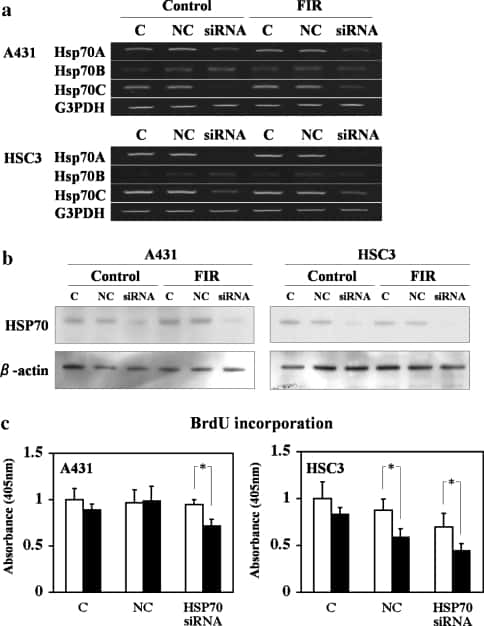 Fig. 4
Open in a separate window
Inhibition of HSP70 expression and BrdU incorporation in A431 and HSC3 cells after 48 h of transfection with 100 nM HSP70 siRNA. (a) RT-PCR of HSP70A, HSP70B, and HSP70C in A431 and HSC3 cells after treatment with siRNAs. C, wild-type control cells; NC, cells transfected with negative control siRNA; siRNA, cells transfected with HSP70-siRNA. (b) Western blot analysis of HSP70 protein expression in A431 and HSC3 cells. The lanes are as shown in panel A. (c) Inhibition of HSP70 suppressed FIR-induced DNA synthesis in A431 cells. Cells were transfected with 100 nM HSP70 siRNA and grown for 48 h. Bars represent the mean and error bars the SD (n = 5). *P < 0.05). In HSC3 cells, the random siRNA slightly suppressed DNA synthesis, whereas the HSP70 siRNA strongly suppressed DNA synthesis
Fig. 4
Open in a separate window
Inhibition of HSP70 expression and BrdU incorporation in A431 and HSC3 cells after 48 h of transfection with 100 nM HSP70 siRNA. (a) RT-PCR of HSP70A, HSP70B, and HSP70C in A431 and HSC3 cells after treatment with siRNAs. C, wild-type control cells; NC, cells transfected with negative control siRNA; siRNA, cells transfected with HSP70-siRNA. (b) Western blot analysis of HSP70 protein expression in A431 and HSC3 cells. The lanes are as shown in panel A. (c) Inhibition of HSP70 suppressed FIR-induced DNA synthesis in A431 cells. Cells were transfected with 100 nM HSP70 siRNA and grown for 48 h. Bars represent the mean and error bars the SD (n = 5). *P < 0.05). In HSC3 cells, the random siRNA slightly suppressed DNA synthesis, whereas the HSP70 siRNA strongly suppressed DNA synthesis
Discussion
In the present studies, we showed that FIR suppresses the proliferation of HSC3, Sa3, and A549 cell lines. Two other cell lines, A431 and MCF7, showed almost no growth arrest in response to FIR. The effect of FIR does not seem to be related to the cell type or source, because these three cell lines have different origins: HSC3 is from a tongue squamous cell carcinoma, Sa3 is from a gingival squamous cell carcinoma, and A549 is from a pulmonary adenocarcinoma. FIR was found to cause hypertrophy without apoptosis in all three sensitive cell lines, although there was a slight increase in necrosis in the Sa3 cells by histological observation. In addition, the expression of apoptosis- related genes was unchanged in the FIR-sensitive cells by microarray analysis. Overall, FIR seemed to cause changes in the cytoskeleton, suppress proliferation, and induce some necrosis without apoptosis. These results raise the question of why proliferation of A431 and MCF7 was not suppressed by FIR. Perhaps something in certain cancer cell lines is present for resistance to FIR. We then focused on genes encoding the HSPs, which are well known to participate in the cellular resistance to stress. We found that HSP70 showed the highest correlation with the growth rate of cancer cells affected by FIR in 35 HSP genes on the microarray system. The expression of HSP70A mRNA was higher in FIR-insensitive A431 and MCF7 cell lines than in FIR-sensitive HSC3, Sa3, and A549 cell lines, although the gene expressions were not induced by FIR. This finding was confirmed by analysis of the expression of HSP70 mRNA and protein with real-time reverse transcription-polymerase chain reaction and enzyme-linked immunosorbent assay. These results suggested that the effect on body temperature range by FIR, suppressing the proliferation of some cancer cells, may be controlled by the basal expression level of HSP70A. To test this hypothesis, the effects of overexpression and suppression of the HSP70 gene were studied. As a result, the overexpression of HSP70 inhibited FIR-induced growth arrest in HSC3 cells and an HSP70 siRNA inhibited the proliferation of A431 cells by FIR. These results confirmed that the effect on body temperature range by FIR, suppressing the proliferation of some cancer cells, is controlled by the basal expression level of HSP70A. HSP70 appears to be present in a variety of normal cell types and its expression may be induced by several stressors, such as hyperthermia, cardiac ischemia, infection, UV radiation, endotoxin, and nitric oxide to suppress or denature any foreign protein and restore an injured protein from lethal effects [12]. HSP70 seems to be particularly important for cancer cells. In human breast cancer, the expression of HSP70 correlates with increased cell proliferation, poor differentiation, lymph node metastases, and poor therapeutic outcome [13]. In vivo animal studies and clinical trials have revealed that hyperthermia may serve as a powerful tool in the treatment of prostate cancer [14–19]; at the cellular level, hyperthermic stress induces HSPs. Moreover, chemotherapeutic agents such as cisplatin, adriamycin, and bleomycin, as well as γ-radiation induce HSPs. HSP70 participates in cytoprotection and is associated with cellular resistance to lethal external effects [17–20]. However, in the present study, HSP70 was never induced by FIR. These results suggested that FIR has anti-tumor activity without inducing HSP70 as an anti-stress factor. This characteristic indicates that FIR may be suitable for medical treatment. Neutralization of the function of HSP70 or inhibition of its expression may inhibit tumor growth and/or sensitize tumor cells to chemotherapeutic agents without affecting normal cells. Furthermore, inhibition of HSP70 expression reduces cell survival. Conversely, microinjection of an HSP70-neutralizing antibody increases the vulnerability of cells to damage by sub-lethal temperatures [21, 22]. This finding was consistent with our results for FIR and the suggestion of HSP70 as the acting mechanism. However, FIR may be regarded as a sub-lethal stress that avoids an anti-stress reaction in some cancer cells. This is the first report suggesting that HSP70 may not rescue cells from the effects of FIR, although HSP70 is known to rescue cells from tumor necrosis, factor-induced caspase-independent programed cell death, heat shock, serum starvation, and oxidative stress [23]. This finding suggests that the effect of FIR may be a sub-lethal stress for cancer cells protected by HSP70 as an anti-stress protein. In other words, FIR may be a very effective medical treatment for some cancer cell lines that have a low level of HSP70. Furthermore, if the level of HSP70 in any cancer of a patient is measured, the effect of medical treatment by FIR on the cancer may be predicted.
Conclusion
It was found here for the first time the effect on body temperature range by FIR in several cancer cell lines in vitro, which is controlled by endogenous HSP70 to protect cells from FIR-induced growth arrest. This finding suggests that FIR may be a very effective medical treatment for some cancer cell lines that have a low level of HSP70. Furthermore, if the level of HSP70 in any cancer of a patient is measured, the effect of medical treatment by FIR on the cancer may be predicted.
Acknowledgment
This research was supported in part by the following grants: Ministry of Economy, Trade and Industry, 41104503; Toseki Matera f19008001.
References
1.Udagawa Y, Nagasawa H, Kiyokawa S. Inhibition by whole-body hyperthermia with far-infrared rays of the growth of spontaneous mammary tumours in mice. Anticancer Res 1999;19(5B):4125–30. [PubMed] 2.Udagawa Y, Nagasawa H. Effects of combined treatment with coffee cherry and whole-body hyperthermia on the growth of spontaneous mammary tumours in SHN mice. In vivo 2000;14(3):431–5. [PubMed] 3.Udagawa Y, Inada K, Nagasawa H. Inhibition by single whole-body hyperthermia with glucose administration of the growth of spontaneous mammary tumors in mice. Jpn J Hyperthermic Oncol 2000;16:229–36. 4.Nagasawa H, et al. Different schedules of whole-body hyperthermia with or without glucose for the inhibition of mammary tumors and uterine adenomyosis in SHN mice. Bull Sch Agr, Meiji Univ 2001;127:43–51. 5.Udagawa Y, Nagasawa H. Effects of far-infrared ray on reproduction, growth, behaviour and some physiological parameters in mice. In vivo 2000;14(2):321–6. [PubMed] 6.Nagasawa Y, Udagawa Y, Kiyokawa S. Evidence that irradiation of far- infrared rays inhibits mammary tumour growth in SHN mice. Anticancer Res 1999;19(3A):1797–800. [PubMed] 7.Inoue S, Kabaya M. Biological activities caused by far-infrared radiation. Int J Biometeorol 1989;33(3):145–50. [PubMed] 8.Honda K, Inoue S. Sleep-enhancing effects of far-infrared radiation in rats. Int J Biometeorol 1988;32(2):92–4. [PubMed] 9.Hosokawa H, et al. A new animal raiser : effect of limited infrared radiation on tumor growth of A431 cells. ITE Lett 2005;6(6):597–602. 10.Teraoka F, Hamada Y, Takahashi J. Bamboo charcoal inhibits growth of HeLa cells in vitro. Dent Mater J 2004;23(4):633–7. [PubMed] 11.Yamashita K, et al. Development of CO2 incubator with limited far-infrared radiation for activation of glucose metabolism. ITE Lett 2005;6(5):53–7. 12.Wong HR, et al. Increased expression of heat shock protein-70 protects A549 cells against hyperoxia. Am J Physiol 1998;275(4 Pt 1):L836–41. [PubMed] 13.Nylandsted J, Brand K, Jaattela M. Heat shock protein 70 is required for the survival of cancer cells. Ann N Y Acad Sci 2000;926:122–5. [PubMed] 14.Kaplan I, Kapp DS, Bagshaw MA. Secondary external-beam radiotherapy and hyperthermia for local recurrence after 125-iodine implantation in adenocarcinoma of the prostate. Int J Radiat Oncol Biol Phys 1991;20(3):551– 4. [PubMed] 15.Kaver I, Ware JL, Koontz WW Jr. The effect of hyperthermia on human prostatic carcinoma cell lines: evaluation in vitro. J Urol 1989;141(4):1025–7. [PubMed] 16.Paulus JA, et al. Heat shock protein response in a prostate tumor model to interstitial thermotherapy: implications for clinical treatment. Prostate 1993;23(3):263–70. [PubMed] 17.Roigas J, et al. Effects of combined treatment of chemotherapeutics and hyperthermia on survival and the regulation of heat shock proteins in Dunning R3327 prostate carcinoma cells. Prostate 1998;34(3):195–202. [PubMed] 18.Servadio C, Leib Z. Local hyperthermia for prostate cancer. Urology 1991;38(4):307–9. [PubMed] 19.Yerushalmi A. Localized, non-invasive deep microwave hyperthermia for the treatment of prostatic tumors: the first 5 years. Recent Results Cancer Res 1988;107:141–6. [PubMed] 20.Bellmann K, et al. Heat shock protein hsp70 over expression confers resistance against nitric oxide. FEBS Lett 1996;391(1–2):185–8. [PubMed] 21.Kiang JG, Tsokos GC. Heat shock protein 70 kDa: molecular biology, biochemistry, and physiology. Pharmacol Ther 1998;80(2):183–201. [PubMed] 22.Riabowol KT, Mizzen LA, Welch WJ. Heat shock is lethal to fibroblasts microinjected with antibodies against hsp70. Science 1988;242(4877):433–6. [PubMed] 23.Vargas-Roig LM, et al. Heat shock proteins and cell proliferation in human breast cancer biopsy samples. Cancer Detect Prev 1997;21(5):441–51. [PubMed]
